Informatics Challenges in Biotherapeutic Glycan Characterization
July 19, 2021
Glycoproteins are now of prime interest to the biopharmaceutical industry, and are being approved for extensive clinical exploration. The qualitative and quantitative existence of glycans in a protein is crucial because the glycosylation (one of the most common post translational modifications) is found to have significant impact on physicochemical properties and thermal stability of a protein molecule. This, in turn, affects the safety and efficacy of glycoprotein-based drugs. Glycans, as we all know, exist either in a free state or in a covalently linked form such as glycopeptides, glycolipids etc. To study and reveal the therapeutic importance of glycans, it is important to release glycans from the protein. It is a multi-step process where a glycoprotein is enzymatically (using enzymes such as Trypsin, Chymotrypsin etc.) cleaved to produce glycopeptides. These glycopeptides are further deglycosylated to release the glycans. Although there are several methods available, enzymatic deglycosylation using PNGaseF is the most popular method.
Glycan structures are diverse and complex because of i) various branching patterns, ii) different monosaccharide residues, iii) topological differences. For detailed elucidation of these structures (including the isomeric glycan structures), MS, MS/MS, and higher tandem MS are required. However, the biological systems often do not produce the samples in an adequate amount. Hence the importance of separation techniques such as Liquid Chromatography (LC) in conjunction with Mass Spectrometry (MS) was felt. Liquid chromatography separates pools of glycans and mass spectrometry helps identify the glycan structures. However, manual processing of these are extremely resource and time intensive, which is why the biotherapeutic industry is in need of an informatics system that addresses the following challenges:
- Baseline Noise: The output of the LC column always suffers with noise because of the column condition. Signal to noise ratio (SNR) of a peak determines whether the peak has to be picked or rejected. To pick the valid peaks from an LC run in a high throughput manner, it is very important to decide a baseline noise level.
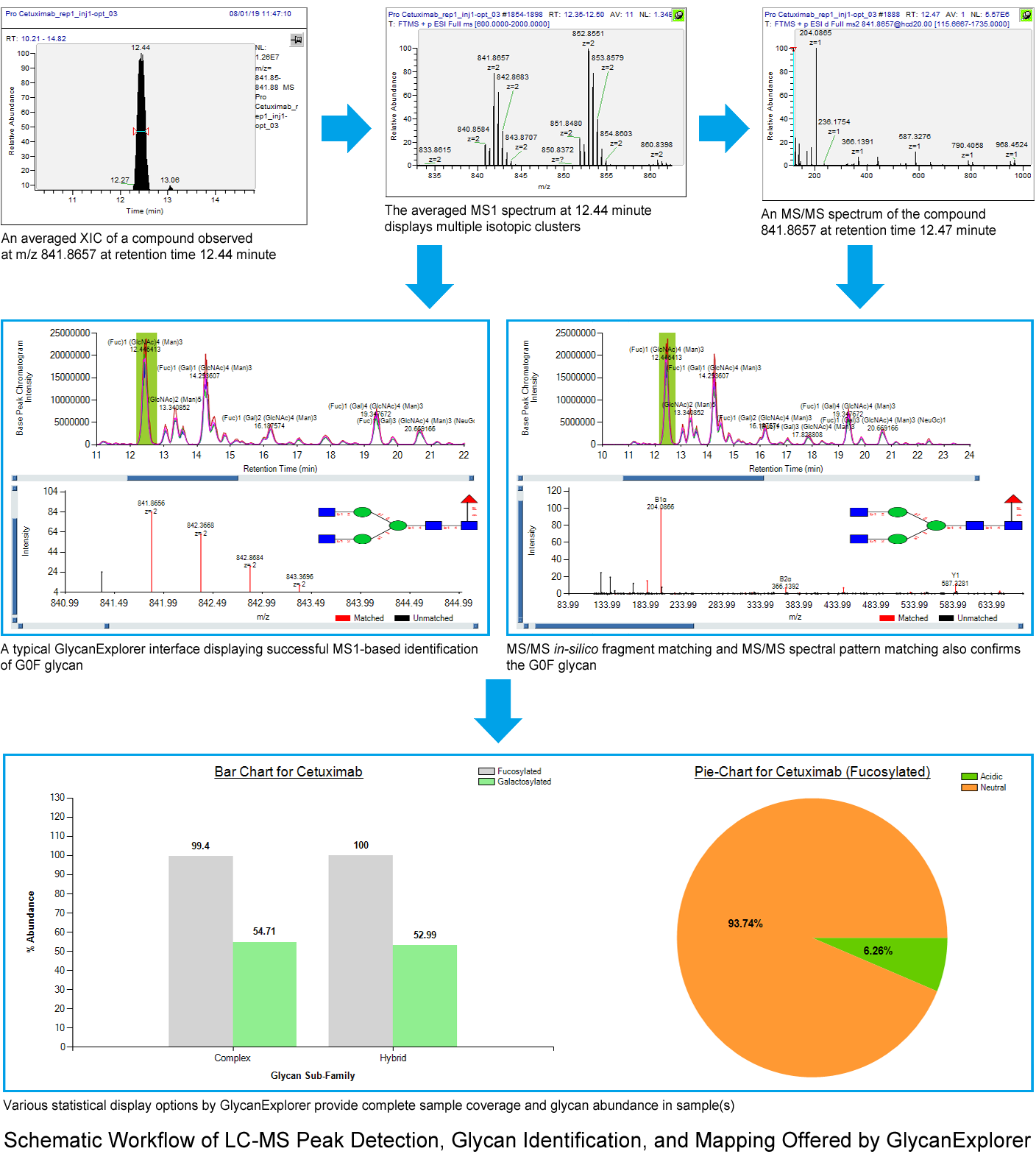
- Adduct Identification: In LC-ESI-MS method, the glycan ions mostly observed are protonated ions, alkali metal ions or ammonium ions. An MS1 spectrum or an average MS1 spectrum of a compound may display multiple isotopic clusters. These clusters may represent:
a. The same compound with same adduct combination, however, at different charge states, or
b. The same compound with different adduct combination and charge states
The informatics solution must have the capability to read the clusters intelligently and assign correct ion species information to the compound. It further helps in accurate glycan identification. - Calculation of Peak Size: In chromatography, various components are often sufficiently separated so that each one displays distinct peaks defined by a specific retention start time and end time. Calculation of peak size is important for quantitative analysis in biotherapeutic research. There are several different measurements such as, peak width, height, and area to calculate the size of a peak. However, peak height and width may suffer from peak broadening issues, and hence, calculating the peak area is always preferred.
- Separation of Co-eluting Isomeric Peaks: One of the very challenging tasks is separating the peaks of isomeric compounds, as the chromatography cannot separate them distinctively because of the same mass. Informatics solution providers nowadays rely on various statistical models such as Savitzky-Golay, Local Minima etc. to resolve these co-eluting peaks and use tandem MS data to identify the correct glycan structure.
- Low Abundant Peaks: As mentioned above, the glycan moieties attached to the proteins can directly affect their thermal stability and eventually the efficacy of glycoprotein-based drugs. Therefore, the functioning of all different glycans of a glycoprotein must be thoroughly analyzed. The structural complexity of glycans, peak broadening, and poor ionization efficiency during liquid chromatography sometimes results in low abundant peaks. The bio-therapeutic industries can not afford to overlook these compounds. There has to be a way to automatically detect these compounds and identify those.
Another way to detect the low abundant peaks is sample derivatization prior to LC-MS analysis. There are many derivatization methods such as permethylation, derivatization using a fluorophore etc. - High Throughput Analysis Capabilities: Bio-therapeutic research demands efficiency in terms of accuracy and time. MS1-based glycan identification protocol has gained popularity because most of the therapeutic glycoproteins of interest are pre-characterized. Therefore, an exact mass search using the appropriate ion species combination and retention time suffice the industry requirements. However, MS1 data fails to distinguish between different isomeric glycan structures (structural isomers and positional isomers). MS/MS-based identification strategies are helpful in identifying the accurate glycan structures.
GlycanExplorer software for biotherapeutic glycan characterization accommodates a fast and robust LC-MS peak picking algorithm that accurately detects and picks the compounds from a chromatographic run by removing the baseline noise, separating the co-eluting isomers, and verifying the acceptable isotopic cluster patterns. The software also employs a proprietary ion alignment algorithm that scans all compounds from all technical replicates of a biological sample, and picks the valid compounds only. The software allows using both MS1 and MS/MS-based approaches (such as, MS/MS in-silico fragment matching and MS/MS spectral pattern matching) for high confidence glycan identification. Using the peak area, the interactive charts and tables efficiently demonstrate the intra-sample as well as inter-sample quantitative comparison (% of fucosylation, % of sialylation).
| Comment | Share |
|


