References >> Mass Spectrometry
Mass SpectrometryMass spectrometry is a powerful analytical technique used to quantify known materials, to identify unknown compounds within a sample, and to elucidate the structure and chemical properties of different molecules. The complete process involves the conversion of the sample into gaseous ions, with or without fragmentation, which are then characterized by their mass to charge ratios (m/z) and relative abundances.
This technique basically studies the effect of ionizing energy on molecules. It depends upon chemical reactions in the gas phase in which sample molecules are consumed during the formation of ionic and neutral species.
Basic Principle
A mass spectrometer generates multiple ions from the sample under investigation, it then separates them according to their specific mass-to-charge ratio (m/z), and then records the relative abundance of each ion type.
The first step in the mass spectrometric analysis of compounds is the production of gas phase ions of the compound, basically by electron ionization. This molecular ion undergoes fragmentation. Each primary product ion derived from the molecular ion, in turn, undergoes fragmentation, and so on. The ions are separated in the mass spectrometer according to their mass-to-charge ratio, and are detected in proportion to their abundance. A mass spectrum of the molecule is thus produced. It displays the result in the form of a plot of ion abundance versus mass-to-charge ratio. Ions provide information concerning the nature and the structure of their precursor molecule. In the spectrum of a pure compound, the molecular ion, if present, appears at the highest value of m/z (followed by ions containing heavier isotopes) and gives the molecular mass of the compound.
Components
The instrument consists of three major components:
-
Ion Source: For producing gaseous ions from the substance being studied.
-
Analyzer: For resolving the ions into their characteristics mass components according to their mass-to-charge ratio.
-
Detector System: For detecting the ions and recording the relative abundance of each of the resolved ionic species.
In addition, a sample introduction system is necessary to admit the samples to be studied to the ion source while maintaining the high vacuum requirements (~10-6 to 10-8 mm of mercury) of the technique; and a computer is required to control the instrument, acquire and manipulate data, and compare spectra to reference libraries.
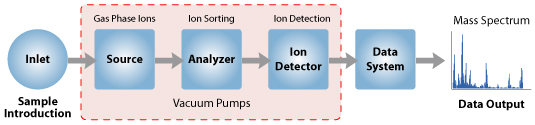
With all the above components, a mass spectrometer should always perform the following processes:
-
Produce ions from the sample in the ionization source.
-
Separate these ions according to their mass-to-charge ratio in the mass analyzer.
-
Eventually, fragment the selected ions and analyze the fragments in a second analyzer.
-
Detect the ions emerging from the last analyzer and measure their abundance with the detector that converts the ions into electrical signals.
-
Process the signals from the detector that are transmitted to the computer and control the instrument using feedback.
Analysis of Biomolecules using Mass Spectrometry
Mass spectrometry is fast becoming an indispensable field for analyzing biomolecules. Till the1970s, the only analytical techniques which provided similar information were electrophoretic, chromatographic or ultracentrifugation methods. The results were not absolute as they were based on characteristics other than the molecular weight. Thus the only possibility of knowing the exact molecular weight of a macromolecule remained its calculation based on its chemical structure.
The development of desorption ionization methods based on the emission of pre-existing ions such as plasma desorption (PD), fast atom bombardment (FAB) or laser desorption (LD), allowed the application of mass spectrometry for analyzing complex biomolecules.
Analysis of Glycans
Oligosaccharides are molecules formed by the association of several monosaccharides
linked through glycosidic bonds. The determination of the complete structure of oligosaccharides is more complex than that of proteins or oligonucleotides. It involves the determination of additional components as a consequence of the isomeric nature of monosaccharides and their capacity to form linear or branched oligosaccharides. Knowing the structure of an oligosaccharide requires not only the determination of its monosaccharide sequence and its branching pattern, but also the isomer position and the anomeric configuration of each of its glycosidic bonds.
Advances in glycobiology involves a comprehensive study of structure, bio-synthesis, and biology of sugars and saccharides. Mass spectrometry (MS) is emerging as an enabling technology in the field of glycomics and glycobiology.
Analysis of Lipids
Lipids are made up of many classes of different molecules which are soluble in organic solvents. Lipidomics, a major part of metabolomics, constitutes the detailed analysis and global characterization, both spatial and temporal, of the structure and function of lipids (the lipidome) within a living system.
Many new strategies for mass-spectrometry-based analyses of lipids have been developed. The most popular lipidomics methodologies involve electrospray ionization (ESI) sources and triple quadrupole analyzers. Using mass spectrometry, it is possible to determine the molecular weight, elemental composition, the position of branching and nature of substituents in the lipid structure.
Analysis of Proteins and Peptides
Proteins and peptides are linear polymers made up of combinations of the 20 amino acids linked by peptide bonds. Proteins undergo several post translational modifications, extending the range of their function via such modifications.
The term Proteomics refers to the analysis of complete protein content in a living system, including co- and post-translationally modified proteins and alternatively spliced variants. Mass Spectrometry has now become a crucial technique for almost all proteomics experiments. It allows precise determination of the molecular mass of peptides as well as their sequences. This information can very well be used for protein identification, de novo sequencing, and identification of post-translational modifications.
Analysis of Oligonucleotides
Oligonucleotides (DNA or RNA), are linear polymers of nucleotides. These are composed of a nitrogenous base, a ribose sugar and a phosphate group. Oligonucleotides may undergo several natural covalent modifications which are commonly present in tRNA and rRNA, or unnatural ones resulting from reactions with exogenous compounds. Mass spectrometry plays an important role in identifying these modifications and determining their structure as well as their position in the oligonucleotide. It not only allows determination of the molecular weight of oligonucleotides, but also in a direct or indirect manner, the determination of their sequences.
Software for Mass Spectrometric Data Analysis
SimGlycan® predicts the structure of glycans and glycopeptides from the MS/MS data acquired by mass spectrometry, facilitating glycosylation and post translational modification studies. SimGlycan® accepts the experimental MS profiles, of both glycopeptides and released glycans, matches them with its own database and generates a list of probable structures. The software also supports multi stage mass spectrometry data analysis which enables structural elucidation and identification of fragmentation pathways.
SimLipid is an innovative lipid characterization tool which enables structural elucidation of unknown lipids using MS/MS data. The software analyzes lipid mass spectrometric data for characterizing and profiling lipids. SimLipid can also annotate mass spectra with the lipid structures identified using abbreviations.






